1.発表者:
栗原 由紀子(東京大学大学院医学系研究科 分子細胞生物学専攻 代謝生理化学分野 講師)
栗原 裕基 (東京大学大学院医学系研究科 分子細胞生物学専攻 代謝生理化学分野 教授)
浴本 亨 (横浜市立大学大学院生命医科学研究科 生命医科学専攻 助教)
池口 満徳 (横浜市立大学大学院生命医科学研究科 生命医科学専攻 教授)
Christopher T. Gordon (INSERM;Institut Imagine and Université Paris-Cité 研究員)
Jeanne Amiel (INSERM;Institut Imagine and Université Paris-Cité,
Hospital Necker-Enfants Malades チームリーダー)
2.発表のポイント:
- 先天性希少疾患である「脱毛症を伴う顎顔面骨形成不全症」を示すエンドセリンA受容体(Gタンパク質共役受容体;GPCRの1つ)変異症例の発症メカニズムを、モデルマウス、薬理実験、分子動力学(MD)シミュレーションを駆使して解明しました。
- 受容体の1アミノ酸変異によって、生体において正常ではほとんど結合しないリガンドが強く結合することでヒトの症状が発症することを証明しました。そしてそのメカニズムが、リガンド結合部位から離れている変異がアロステリックにリガンド結合に影響を与え、リガンドの乖離を阻害し結合性を増加させることを明らかにしました。
- 今回解明したエンドセリンA受容体変異の発症機構は、多くのGPCRの活性化に共通するメカニズムと考えられます。すなわち、細胞のシグナル伝達を担う主要な受容体であるGPCRは薬のターゲットの1/3以上を占めていることから、GPCR活性化機序を詳細に明らかにしたことは創薬のデザインにも大きく貢献すると考えられます。
3.発表概要:
特定の遺伝子異常が原因の希少疾患はいろいろ知られていますが、その発症メカニズムの多くは解明されていません。今回、東京大学大学院医学系研究科代謝生理化学分野 栗原由紀子講師らのグループと、INSERM(フランス国立衛生医学研究所)、横浜市立大学大学院生命医科学研究科との共同研究で、原子のレベルから顎顔面骨形成不全症を示すエンドセリンA受容体変異の発症機序を解明しました。この受容体タンパク質は、ヘリックスの細胞内側の1つのアミノ酸の荷電が変化して周囲との水素結合が失われると大きく動くようになり、ヘリックスの細胞外側のリガンド結合部位の構造に影響を与え、新たな機能を獲得することがわかりました。ヒト希少疾患の発症機構について、遺伝子改変マウスや薬理の実験系とスーパーコンピューターを用いた理論系をうまく組み合わせて理解できた良い成功例となりました。
4.発表内容:
我々はこれまで、エンドセリン1(注1)やエンドセリンA受容体(ETAR)の遺伝子改変マウスにより、エンドセリン1―エンドセリンA受容体シグナルが頭部神経堤細胞に作用して、頭部顔面や心臓大血管の形成に重要であること、特に顎顔面形成においては、エンドセリンシグナルが核転写因子Dlx5/6の誘導を介して上顎下顎を決定することを明らかにしてきました。一方、パリのネッカー小児病院のJeanne Amielチームリーダーらの研究グループは、耳介、顔面の形態異常や難聴を示す「脱毛症を伴う顎顔面骨形成不全症」の症例の中に、エンドセリンA受容体の1塩基変異:G→A、アミノ酸ではグルタミン酸からリジンへの変異(E303K)があることを次世代シークエンサー等で見出しました(図1)。そこで、本当にこれらの1アミノ酸変異が発症の原因であるかを確かめるために、ヒトと全く同じ1アミノ酸変異だけを持つマウスをCRISPR/CASを用いて作成したところ、耳介の形態異常、上顎骨の形態異常、難聴を推測させる耳小骨の低形成が再現されました(図2)。同時に、我々が以前報告したエンドセリンA受容体Y129F変異(チロシンからフェニルアラニンへの変異)症例のモデルマウスも、ヒト症例を模倣しかつE303K変異マウスとほぼ同様の表現型を示しました。
この上顎骨の異常は、エンドセリンシグナルを異所性に上顎に発現させたマウスの上顎骨に似ていることから、エンドセリンシグナルの機能獲得変異である可能性を考えました。胎生期の将来上顎になる鰓弓部分にはもともとエンドセリン-3が発現していますが、エンドセリン-3は生体ではエンドセリンA受容体にほとんど結合しません。そこで、「エンドセリンA受容体の変異によってエンドセリン-3と結合するようになった」と仮説を立てました。この仮説は、CRISPR/CASで作成したエンドセリン-3ノックアウトによって表現型が正常に戻ったこと(図2)と、培養細胞の系でE303K変異またはY129F変異のエンドセリンA受容体が、エンドセリン-3との親和性を獲得し受容体の下流シグナルが上昇した(図3)ことから証明されました(図4)。
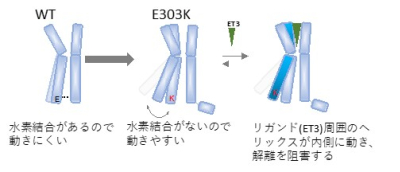
では、リガンド(注2)結合部位のから離れたところにあるE303K, Y129F変異はどのようにしてエンドセリン-3の親和性を増加させたのでしょうか?E303Kでは、グルタミン酸からリジンへの変異によってアミノ酸側鎖の電荷がマイナスからプラスへと性質を大きく変えているので、周囲とのイオン結合や水素結合が変化することが想定されました。そこでエンドセリンA受容体のモデリングを施行後MD(分子動力学)シミュレーションを導入しました(図5)。MDシミュレーションは、タンパク質を形成する原子1つ1つに対して加わる力に基づいてニュートンの運動方程式を立てて、すべての原子に加わる力を一度にスーパーコンピューターで計算することにより、タンパク質分子内のオングストロームレベルの動きを可視化します。1ナノ秒ごとに原子の3次元位置座標が得られるので、E303のアミノ酸側鎖がどの原子とどの程度結合するかや、ヘリックス(注3)がどのくらい動くかを確率論的に計算できます。MDシミュレーションにより、ヘリックスの細胞内側に位置するE303Kは隣のヘリックスと結合できなくなって大きく動くようになり、その分細胞外側は狭まる確率が高くなりました。これにより、エンドセリン-3とエンドセリンA受容体は結合すると乖離しにくくなり、親和性が上昇すると考えられます。即ち、ET-3 + ETAR ⇄ ET-3/ETARの平衡が右に大きく傾くということです(図6)。同様に計算すると、受容体の中心部のナトリウム-水ポケットに位置するY129F変異では(図5)、フェニルアラニンはチロシンのOH基がHに置き換わったものですから、周囲の水分子との水素結合がなくなり、水ポケットが小さくなるのに合わせてリガンド結合部位が狭くなる確率が高くなり、E303Kと同様にエンドセリン-3に対する親和性が上昇したと考えられました(図7)。
エンドセリン受容体はアドレナリン受容体やオピオイド受容体などと同じクラスA GPCR(注4)であり、細胞内側のヘリックスの動きやナトリウム-水ポケットについては共通していると考えられるので、GPCRシグナル伝達の研究やGPCRをターゲットとした創薬において重要な知見を与えたと言えます。
本研究は、文部科学省科研費(16K15254, 19K08534, 22H04991)、創薬等先端技術支援基盤プラットフォーム(BINDS)(JP20am0101109, JP22ama121023)、スーパーコンピューター「富岳」成果創出加速プログラム(hp200129, hp210172)などの支援により実施されました。
5.発表雑誌:
雑誌名:「Journal of Clinical Investigation」(オンライン版:1月13日)
論文タイトル:Mandibulofacial dysostosis with alopecia results from gain-of-ETAR function via allosteric effects on ligand binding
著者:Yukiko Kurihara*, Toru Ekimoto, Christopher T. Gordon, Yasunobu Uchijima, Ryo Sugiyama, Taro Kitazawa, Akiyasu Iwase, Risa Kotani, Rieko Asai, Véronique Pingault, Mitsunori Ikeguchi, Jeanne Amiel, Hiroki Kurihara
DOI番号:
https://doi.org/10.1172/JCI151536
6.注意事項:
日本時間1月14日(土)午前2時(米国東部時間:13日(金)正午)以前の公表は禁じられています。
7.用語解説:
(注意1)エンドセリン(Endothelin: ET)
エンドセリンファミリーはET1、2、3の3つのETとETA、ETB受容体2つの受容体からなる。ET-1は血管収縮を主な作用とする生理活性ペプチドで、その受容体拮抗薬は難病の一つである肺動脈性肺高血圧症の治療に使われている。胎生期には顎顔面の形成に重要な役割を果たしている。正常な生体では、ET3はETA受容体をほとんど活性化しない。
(注意2)リガンド
特定の受容体に特異的に結合するタンパク質・ペプチドや低分子化合物。本研究においては、リガンドはエンドセリンであり、受容体との結合能を親和性という。
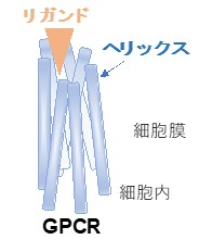
(注意3)ヘリックス(αヘリックス)
蛋白の2次構造で、3.6アミノ酸残基ごとに1回転するらせん構造をとる。GPCRでは7回の細胞膜貫通ドメインがこの構造で作られている。
(注意4)Gタンパク質共役受容体(GPCR)
細胞膜を7回貫通するαヘリックス構造を特徴とする細胞膜受容体。細胞外側でリガンドが結合すると、細胞内側で結合する三量体Gタンパク質が活性化されてシグナルが伝達される。ヒトのゲノムには800種類以上のGPCRがコードされていると言われ、創薬の重要なターゲットにもなっている。
8.添付資料:
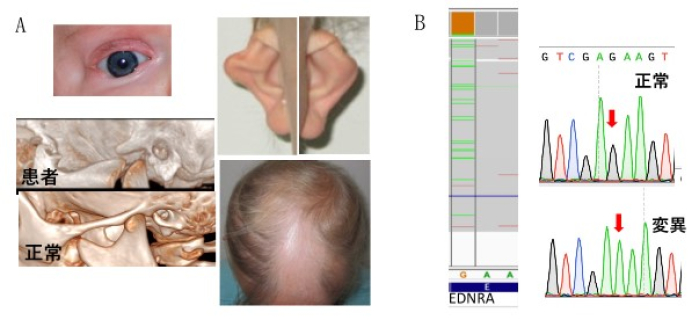
図1 脱毛を伴う顎顔面骨形成不全症(Mandibulofacial Dysostosis with alopecia; MFDA)
A.患児は8歳女性。耳介、上顎骨・顔面の骨の形態異常、脱毛、眼瞼欠損の他、耳小骨の低形成・
難聴、低身長などが見られる。
B.次世代シークエンサーやサンガーシークエンスにより、エンドセリンA受容体(ETAR/EDNRA)に
おけるゲノムDNA 907 G→A変異(アミノ酸E303K)が見つかった。
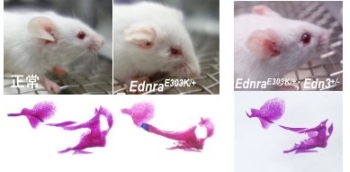
エンドセリン3欠損で回復!
(左・中央)エンドセリンA受容体(ETAR/Ednra)正常マウスとE303K変異マウス。
E303Kでは耳介が折れ曲がり上顎を形成する骨が下顎のように太くなり関節軟骨が形成されている。(紫は骨、青は軟骨を示す。)
(右)ETAR_E303K変異に加えエンドセリン3 (ET3/Edn3 )を半分欠損させたところ、耳介や、上顎の骨が正常になった。
* ETAR_Y129F変異マウスでも同様の表現型が見られ、ET3欠損で正常に回復した。
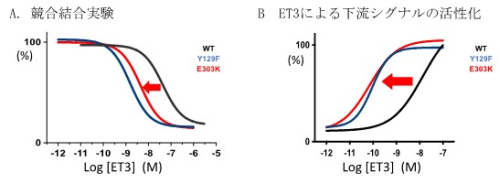
図3 薬理学的なエンドセリン3の親和性上昇
A.培養細胞でET3とETARの結合能(親和性)を評価したところ、Y129F, E303K変異は共に結合能が
上昇した。
B.ETARの下流のシグナルの1つERKのリン酸化能も Y129F, E303K変異で上昇した。
即ち、ET3は胎児に存在する濃度ではETARに結合できなかったが、ETARが変異したことにより生体
でも結合できるようになった。
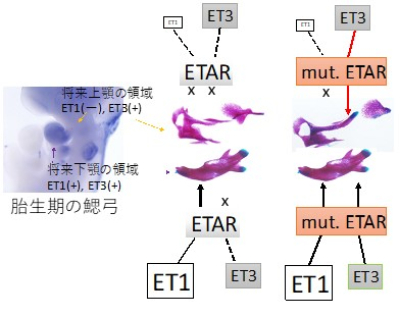
図4 症状出現の機序
正常の上顎では、ET1はほとんど発現していないのでETARを活性化できず、ET3はETARに結合できないのでETARシグナルは発生しない。
一方、変異ETARはET3と十分結合できるので、上顎でシグナルが活性化して、関節軟骨を持つ下顎の様な骨になった。 (紫は骨、青は軟骨。)
*なぜ、リガンド結合ポケットから遠く離れたところに存在するアミノ酸変異で結合性が上昇するのか? →MDシミュレーションで解析
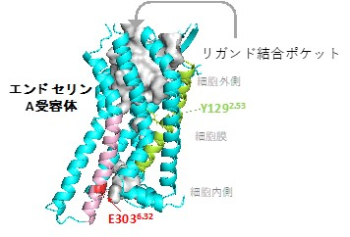
図5 エンドセリンA受容体のモデル
7本のヘリックスに囲まれた上部の灰色の部分がリガンド結合ポケット。
E303Kはヘリックスの細胞内側に存在しているので、リガンド結合に直接には関われない。
Y129Fはリガンド結合ポケットの下のNa-水ポケットに存在する。
図6 ETAR_E303KでET3リガンドの結合性が上昇する理由
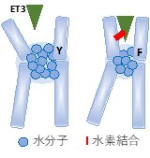
図7 Y129F_ETARでET3リガンドの結合性が上昇する理由
MDシミュレーションを行うことによって、ETAR_Y129FではNa-水ポケットの水分子の数が減りリガンド結合ポケット周囲のヘリックスが内側に動き水素結合が形成されることがわかった。これにより解離を阻害し、リガンド結合性が増すと考えられた。