東京医科大学(学長:林 由起子/東京都新宿区)医学総合研究所未来医療研究センター実験病理学部門の中村卓郎特任教授らが、公益財団法人がん研究会がん研究所がんエピゲノムプロジェクト 田中美和主任研究員、丸山玲緒プロジェクトリーダー、がん研有明病院整形外科 阿江啓介部長、松本誠一サルコーマセンター長、京都大学大学院工学研究科マイクロエンジニアリング専攻 横川隆司教授らとの共同研究で、胞巣状軟部肉腫(alveolar soft part sarcoma, ASPS)の原因融合遺伝子ASPSCR1::TFE3(AT3)の機能とその標的遺伝子を明らかにし、ASPSの血管形成を誘導する仕組みを解明しました。
ASPSは希少がんである軟部肉腫の一つで、AYA世代(思春期・若年成人)に好発します。腫瘍の増殖は緩やかですが、血管形成が盛んなことから全身に転移する傾向が強く、予後不良な疾患です。ASPSの原因融合遺伝子AT3が血管形成をはじめとする腫瘍の形質をコントロールしていることが示唆されていました。本研究グループは2017年にASPSのマウスモデルを確立して研究を進めていましたが、今回AT3蛋白質が血管形成を促進する遺伝子のスーパーエンハンサーに結合することを発見し、エピゲノム編集技術を用いて標的遺伝子を同定しました。標的遺伝子には血管形成因子自体と、それらを運ぶ細胞内輸送促進因子が含まれ、ASPSにおける独特な血管構造の原因となっていることがわかりました。今後、輸送促進因子機能を抑える全く新しい治療方法の開発にもつながる成果と期待されます。
この研究成果は、2023年4月7日(米国時間)のNature Communications誌(オンライン版)に掲載されました。
【本研究のポイント】
●ASPSモデルマウスから樹立した腫瘍細胞株を継代培養すると、融合遺伝子AT3の発現がしばしば消失し、培養条件下では融合遺伝子は不要であることが示唆された。
●AT3を失った細胞をマウスに移植すると、血管形成が消失し腫瘍増殖が著しく抑制された。
●AT3はスーパーエンハンサーを含むアクティブエンハンサーに結合する。
●AT3の発現が消失した腫瘍細胞では血管形成に関連した遺伝子のエンハンサー機能が低下し、遺伝子発現も抑制された。
●エピゲノム編集技術を用いてASPSの血管形成に関わるエンハンサーと標的遺伝子Rab27a, Sytl2, Pdgfb, Vwfを同定した。
●Rab27aとSytl2は細胞内小胞輸送に関わり、血管形成因子の輸送を促進することがマイクロ流体デバイスを用いた解析から証明された。
【研究の背景】
がんの成長にしたがってがん巣内での血管形成が必要になってきます。正常の血管では血管内皮細胞が周皮細胞に裏打ちされた構造をとりますが、がんの新生血管ではしばしば周皮細胞が欠如し、漏れが生じやすい不完全な血管形成が生じます。一方、がんの中にはより完全な血管構造が豊富に存在するタイプがあり、これらのがんは血行性転移が頻発する傾向を示します。ASPSはAYA世代の大腿や臀部、上腕といった深部の軟部組織に発生する悪性の肉腫で、ゆっくりとした発育態度を示します。その一方で、胞巣状構造と呼ばれる腫瘍細胞と周皮細胞に富む血管組織が一体化した病巣を形成するため、比較的早期から高頻度で肺などへの転移を示します。効果的な治療薬が未開発であり、予後不良な疾患です。ASPSはその発生起源が不明な謎の多い腫瘍でもあります。
研究チームでは、ASPSの原因遺伝子であるAT3をマウス胎児の間葉系細胞に導入したモデルを作製しましたが、このモデルではASPSに見られる胞巣状構造と肺転移が忠実に再現され、ASPSの特性の理解に役立ってきました。さらに血管形成機構や融合遺伝子機能、標的遺伝子の解明への貢献が期待されていました。
【本研究で得られた結果・知見】
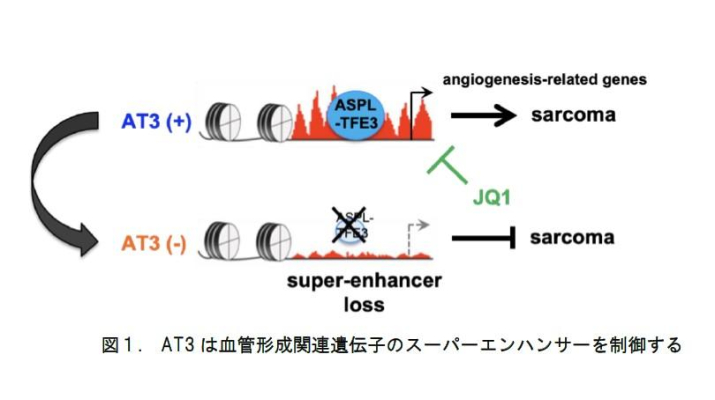
ASPSモデルマウスから樹立した腫瘍細胞株を継代培養していると、融合遺伝子AT3の発現がしばしば消失することから、培養条件下では融合遺伝子注1は不要であることが示唆されました。ところが、AT3を失った細胞をマウスに移植すると、血管形成が消失し腫瘍増殖が著しく抑制されることがわかりました。AT3は、転写因子としてスーパーエンハンサー(注2)を含むアクティブなエンハンサーに結合します。AT3の発現が消失した腫瘍細胞では血管形成に関連した遺伝子のエンハンサー機能が低下し、遺伝子発現も抑制される傾向が認められました(図1)。興味深いことに、スーパーエンハンサーの機能を抑えるBRD4阻害薬JQ1をASPS移植マウスに投与しても同様の効果が認められました。
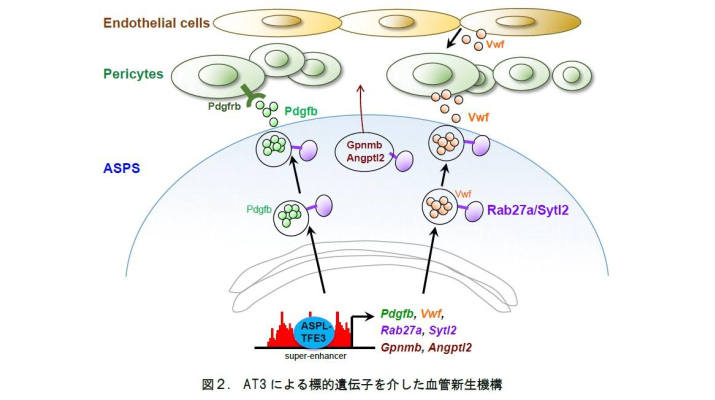
そこで、血管新生と生体内での腫瘍形成に関わるエンハンサーを同定する目的で、CRISPRエピゲノム編集技術注3を利用したスクリーニングを実施すると、Rab27a、Sytl2、Pdgfb、Vwfの4遺伝子がAT3に制御されるエンハンサーに支配されていることがわかりました。この内、Pdgfbは周皮細胞を誘引する因子として、Vwfは血管増殖因子の機能を維持する因子として機能することが示唆されています。一方で、Rab27aとSytl2は血管形成因子を含む小胞の細胞膜への輸送を促進することが知られています。今回の発見はAT3が血管形成因子の産生と分泌という血管形成を統括する機能を有することを示すとともに、がんにおける細胞内輸送経路の重要性を示したものと考えています(図2)。
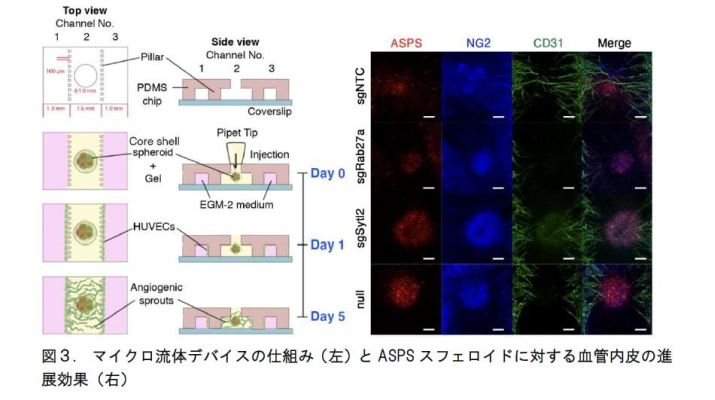
さらに、マイクロ流体デバイス(注4)にASPS細胞と周皮細胞とから成るスフェロイドを細胞外基質と共に導入し、血管内皮細胞の進展を評価しました。スフェロイドをチャネル2に、内皮細胞をチャネル1と3に導入して伸長を観察したところ、血管形成因子Rab27aとSytl2のノックアウトにより血管の伸展が阻害されました。これにより、マウス生体での血管形成因子依存的な血管形成を、マイクロ流体デバイス内で再現することにも成功しました(図3)。
【今後の研究展開および波及効果】
ASPSは患者数も少なく、特異な病像も相まって血管形成機構や転移様式には他の多くのがん(いわゆるコモンキャンサー)とは相違点が少なくありません。しかしながら、ASPSに見られる血管構造は腎細胞がんや肝細胞がん、内分泌系腫瘍などにも認められることから、今回発見された細胞内輸送経路の亢進を介した血管形成の仕組みは、これらのがんにも当てはまることが予想されます。
今回の研究から、AT3融合遺伝子やRab27a/Sytl2の機能を阻害する全く新しい治療薬の開発が大きな意義を持つことがわかり、私たちは創薬研究を開始しています。特にRab27a/Sytl2に促進される小胞輸送をコントロールすることは、がんだけではなく好中球やT細胞が関わる炎症を抑えることもわかっていて、アレルギー疾患や敗血症などへの応用も期待されます。開発したマイクロ流体デバイスについても、創薬研究に向けた実用化を推進していきます。
【掲載誌名・DOI】
掲載誌:Nature Communications
https://www.nature.com/articles/s41467-023-37049-z
DOI:10.1038/s41467-023-37049-z
【論文タイトル】
ASPSCR1::TFE3 orchestrates the angiogenic program of alveolar soft part sarcoma
【著者】
田中美和*、Surachada Chuaychob、本目みずき、山崎ゆかり、Ruyin Lyu、山下享子、阿江啓介、松本誠一、粂川昂平、丸山玲緒、Wei Qu、宮城洋平、横川隆司、中村卓郎*
*責任著者
【主な競争的研究資金】
文部科学省 科学研究費 基盤研究(A) 26250029(中村卓郎)、基盤研究(C) 16KM07131、19K07702(田中美和)
日本医療開発研究機構 次世代がん医療創生研究事業 17cm0106609(中村卓郎)、20cm0106277(中村卓郎、横川隆司)、次世代がん医療加速化研究事業22ama221206(田中美和、横川隆司)
日本医療開発研究機構 創薬基盤推進研究事業 21ak0101170(中村卓郎)
【脚注、用語説明】
注1:融合遺伝子は染色体の構造異常(転座、逆位、欠失など)によって2種類の遺伝子が融合し新たな機能を持つに至った遺伝子で、がんの原因遺伝子として広く認められる。白血病や肉腫で多く、特に肉腫では全体の30%で認められる。慢性骨髄性白血病の融合遺伝子BCR-ABLを標的とするキナーゼ阻害薬は分子標的治療法の先駆的存在だが、肉腫の融合遺伝子の大部分は有効な分子標的薬が未開発で、ASPSにおけるASPSCR1::TFE3も同様である。
注2:エンハンサーはゲノム上の領域の中で、遺伝子をDNAからmRNAへの転写を促進する際に必要とされるもの。転写因子や転写共役因子が複合体を形成して結合し、プロモーター領域のRNAポリメラーゼの機能を促進させる。中でも、スーパーエンハンサーは巨大なエンハンサー領域であり、メディエーター複合体という数多くの転写因子や共役因子が大規模な複合体を形成して、転写を強力に促進させる。スーパーエンハンサーは、細胞の個性を規定するとともに、がんにおける異常な転写活性に関与していることが知られている。
注3:現在ゲノム編集技術の中心的存在であるCRISPR/Cas9は、Cas9によるDNAの二重鎖切断活性を介してゲノムを改変させる。これに対してエピゲノム編集は、変異型Cas9に転写作用を修飾する分子を付加することにより、目的のゲノム領域のエピゲノムを変化させて遺伝子発現の亢進や抑制を狙う。
注4:マイクロ流体デバイス(チップ)は、半導体微細加工技術を用いて流路をガラスや樹脂基板上に作製したものである。臓器細胞を培養して生体内に近い環境を創成したチップは、Microphysiological systems (MPS)あるいは生体模倣システムと呼ばれる。今回の研究では、肉腫・周皮細胞・血管内皮の3種類の細胞を用いてASPSの血管形成を再現させた。
▼本件に関する問い合わせ先
企画部 広報・社会連携推進室
住所:〒160-8402 東京都新宿区新宿6-1-1
TEL:03-3351-6141
メール:d-koho@tokyo-med.ac.jp
【リリース発信元】 大学プレスセンター
https://www.u-presscenter.jp/